## Reference implementation: PairedEndSingleSampleWf pipeline
http://gatkforums.broadinstitute.org/gatk/discussion/7899/reference-implementation-pairedendsinglesamplewf-pipeline
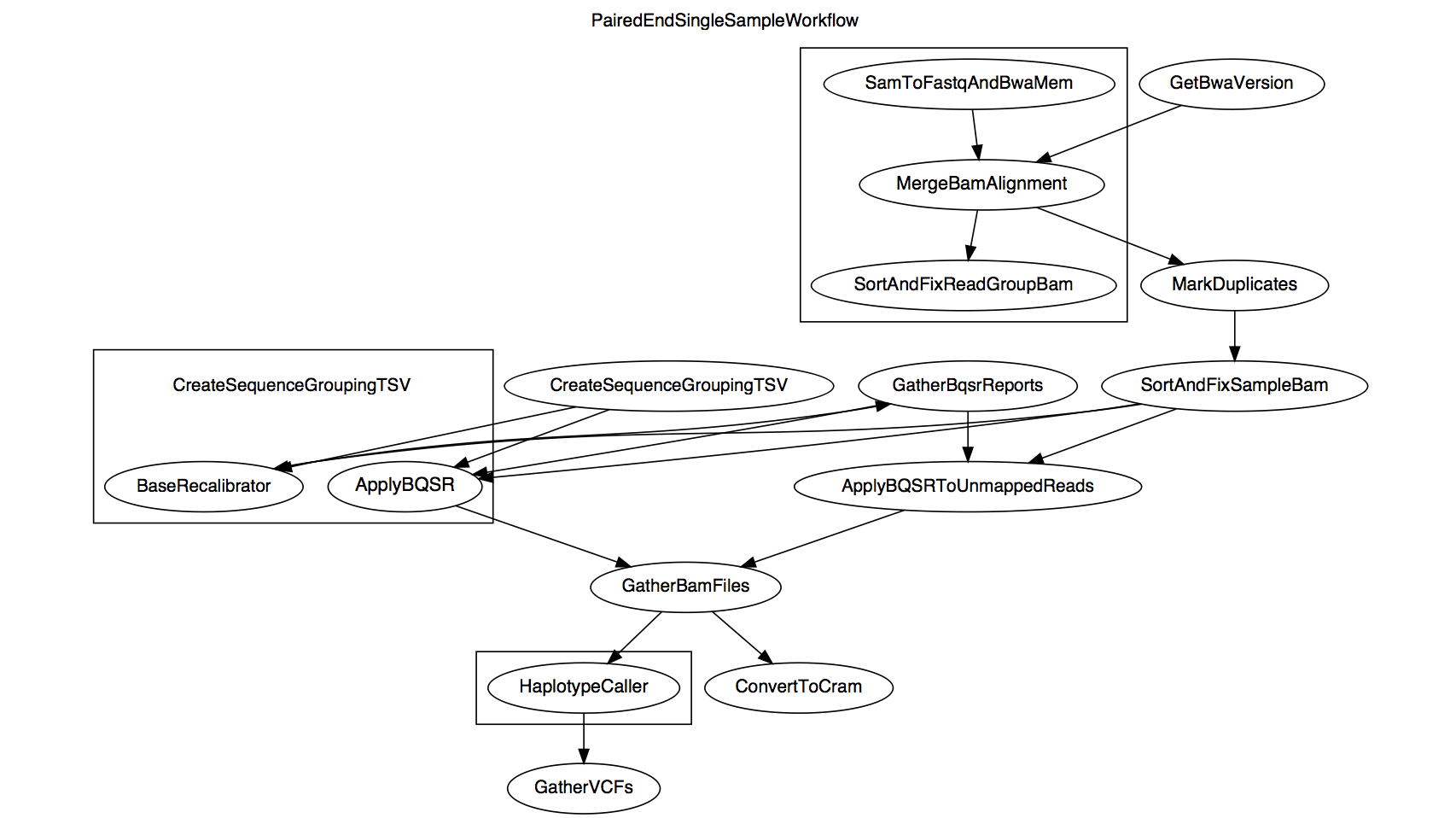 This document describes the workflow and details pertinent parameters of the PairedEndSingleSampleWf pipeline, which implements GATK Best Practices (ca. June 2016) for pre-processing human germline whole-genome sequencing (WGS) data. This pipeline uses GRCh38 as the reference genome and, as the name implies, is specific to processing paired end reads for a single sample. It begins with unaligned paired reads in BAM format and results in a sample-level SNP and INDEL variant callset in GVCF format.
This document describes the workflow and details pertinent parameters of the PairedEndSingleSampleWf pipeline, which implements GATK Best Practices (ca. June 2016) for pre-processing human germline whole-genome sequencing (WGS) data. This pipeline uses GRCh38 as the reference genome and, as the name implies, is specific to processing paired end reads for a single sample. It begins with unaligned paired reads in BAM format and results in a sample-level SNP and INDEL variant callset in GVCF format.
- This document is specific to the public WDL script PublicPairedSingleSampleWf_160720.wdl with a July 20, 2016 date stamp found here.
- The outline uses Docker container broadinstitute/genomes-in-the-cloud:2.2.2-1466113830. Docker containers provide reproducible analysis environments, including specific tool versions. Thus, the commands and the given parameters pertain only to these specific tool versions. It is possible for prior or future versions of tools to give different results.
- Furthermore, the parameters within the pipeline are optimized for WGS and GRCh38. Optimal performance on other types of data and other species may require changes to parameters. For example, Step 3 includes code that calculates how to split the reference contigs for parallel processing that caps the genomic interval per process by the longest contig in the reference. This splits human GRCh38 18-ways and caps each group of contigs by the size of human chromosome 1. This approach may need revision for efficient processing of genomes where the longest contig length deviates greatly from human.
The diagram above shows the relationship between the WORKFLOW steps that call on specific TASKS. Certain steps use genomic intervals to parallelize processes, and these are boxed in the workflow diagram. An overview of the data transformations is given in the WORKFLOW definitions section and granular details are given in the TASK definitions section in the order shown below.
Jump to a section
WORKFLOW definition overview
- Map with BWA-MEM and merge to create clean BAM
- Flag duplicates with MarkDuplicates
- Base quality score recalibration
- Call SNP and INDEL variants with HaplotypeCaller
TASK definitions overview
What is NOT covered
- This document assumes a basic understanding of WDL components.
- The JSON files describing inputs and outputs.
- Runtime parameters optimized for Broad's Google Cloud Platform implementation.
Related resources
Requirements
Software
- See Article#6671 for setup options in using Cromwell to execute WDL workflows. For an introduction, see Article#7349.
- Docker container
broadinstitute/genomes-in-the-cloud:2.2.3-1469027018 uses the following tool versions for this pipeline. These tools in turn require Java JDK v8 (specifically 8u91) and Python v2.7.
DOCKER_VERSION="1.8.1"
PICARD_VERSION="1.1099"
GATK35_VERSION="3.5-0-g36282e4"
GATK4_VERSION="4.alpha-249-g7df4044"
SAMTOOLS_VER="1.3.1"
BWA_VER="0.7.13-r1126"
Scripts and data
- Pipeline WDL script and JSON file defining input data.
- (Optional) JSON file defining additional Cromwell options.
- Human whole genome paired end sequence reads in unmapped BAM (uBAM) format.
- Each uBAM is per read group and the header defines read group
ID, SM, LB, PL and optionally PU. Because each file is for the same sample, their SM fields will be identical. Each read has an RG tag.
- Each uBAM has the same suffix, e.g.
.unmapped.bam.
- Each uBAM passes validation by ValidateSamFile.
- Reads are in query-sorted order.
- GRCh38 reference genome FASTA including alternate contigs, corresponding
.fai index and .dict dictionary and six BWA-specific index files .alt, .sa, .amb, .bwt, .ann and .pac.
- Known variant sites VCFs and corresponding indexes for masking during base quality score recalibration (BQSR). A separate article will document these data resources.
- Intervals lists for scattered variant calling with HaplotypeCaller. It is necessary to predefine the calling intervals carefully. In this workflow we use 50
.interval_list files that each contain multiple calling intervals. The calling intervals are an intersection of (i) calling regions of interest and (ii) regions bounded by Ns, otherwise known as gaps in the genome. See the External Resources section of Article#7857 for an example gap file. Use of these defined intervals has the following benefits.
- Avoids calling twice on a locus. This can happen when reads overlapping an interval boundary expand the interval under consideration such that the same variant is called twice.
- Makes calls taking into consideration all the available reads that align to the same locus.
WORKFLOW definition overview
Below we see that the workflow name is PairedEndSingleSampleWorkflow.
[0.0]

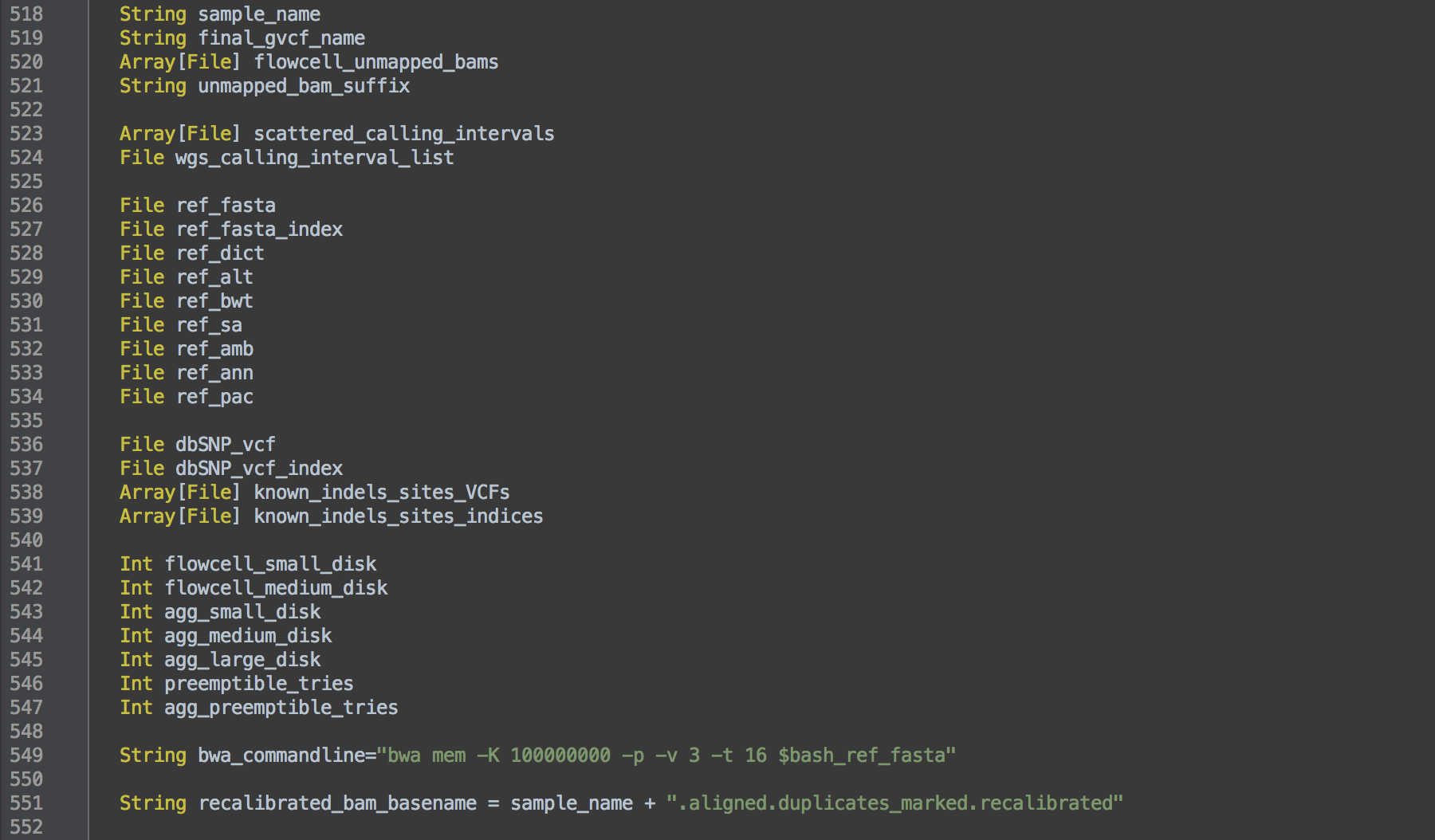
After the workflow name, the WORKFLOW definition lists the variables that can stand in for files, parameters or even parts of commands within tasks, e.g. the command for BWA alignment (L549). The actual files are given in an accompanying JSON file.
[0.1]
The WORKFLOW definition then outlines the tasks that it will perform. Because tasks may be listed in any order, it is the WORKFLOW definition that defines the order in which steps are run.
Let's break down the workflow into steps and examine their component commands.
back to top
1. Map with BWA-MEM and merge to create clean BAM
This step takes the unaligned BAM, aligns with BWA-MEM, merges information between the unaligned and aligned BAM and fixes tags and sorts the BAM.
- 1.0: Calls task GetBwaVersion to note the version of BWA for use in step [1.3].
- 1.1: Defines how to scatter the given unmapped BAM for [1.2], [1.3] and [1.4]. The workflow scatters each
unmapped_bam in the list of BAMs given by the variable flowcell_unmapped_bams. The step processes each unmapped_bam in flowcell_unmapped_bams separately in parallel for the three processes. That is, the workflow processes each read group BAM independently for this step.
▶︎ Observe the nesting of commands via their relative indentation. Our script writers use these indentations not because they make a difference for Cromwell interpretation but because they allow us human readers to visually comprehend where the scattering applies. In box [1.1] below, we see the scattering defined in L558 applies to processes in boxes [1.2], [1.3] and [1.4] in that the script nests, or indents further in, the commands for these processes within the scattering command.
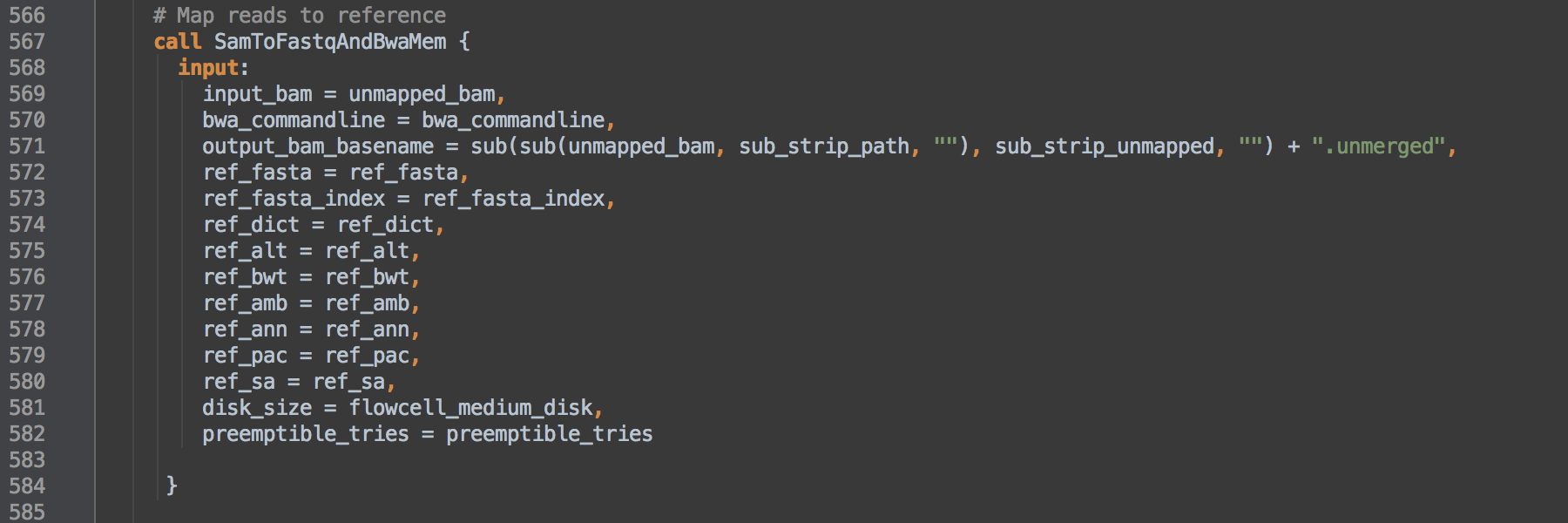
- 1.2: Calls task SamToFastqAndBwaMem to map reads to the reference using BWA-MEM. We use
bwa_commandline from L549 as the actual command.
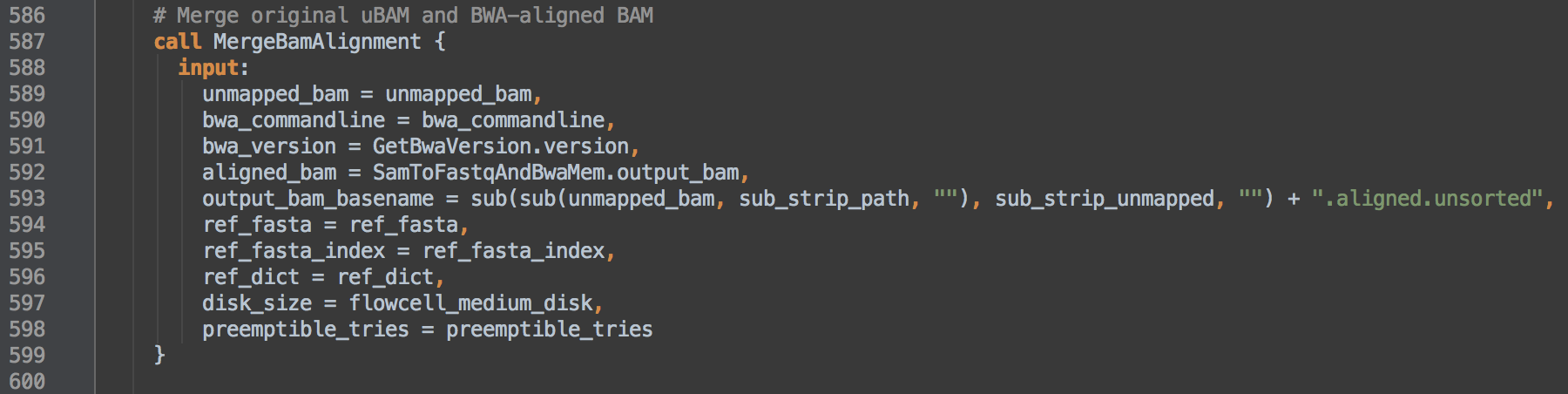
- 1.3: Calls task MergeBamAlignment to merge information between the unaligned and aligned BAM. Both
bwa_commandline and bwa_version define elements of the bwamem program group @PG line in the BAM header. The data resulting from this step go on to step [2].
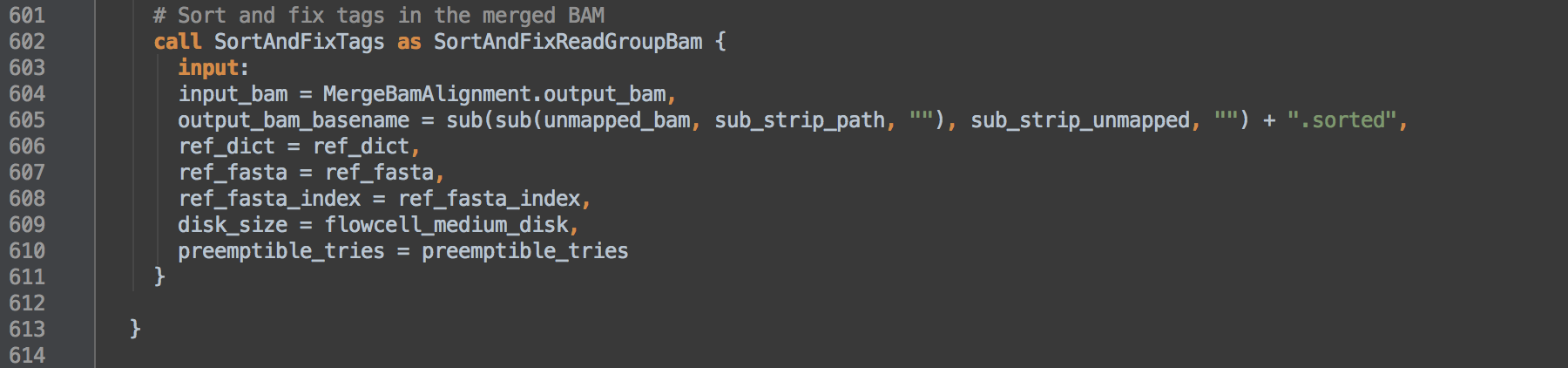
- 1.4: Calls task SortAndFixTags and uses task aliasing to rename it SortAndFixReadGroupBam. The consequence of this is that the workflow can then differentiate outputs from those of [2.1]. This task coordinate sorts and indexes the alignments and fixes the
NM and UQ tags whose calculations depend on coordinate sort order. This data transformation allows for validation with ValidateSamFile.
[1.0]

[1.1]
[1.2]
[1.3]
[1.4]
back to top
2. Flag duplicates with MarkDuplicates
This step aggregates sample BAMs, flags duplicate sets, fixes tags and coordinate sorts. It starts with the output of [1.3]
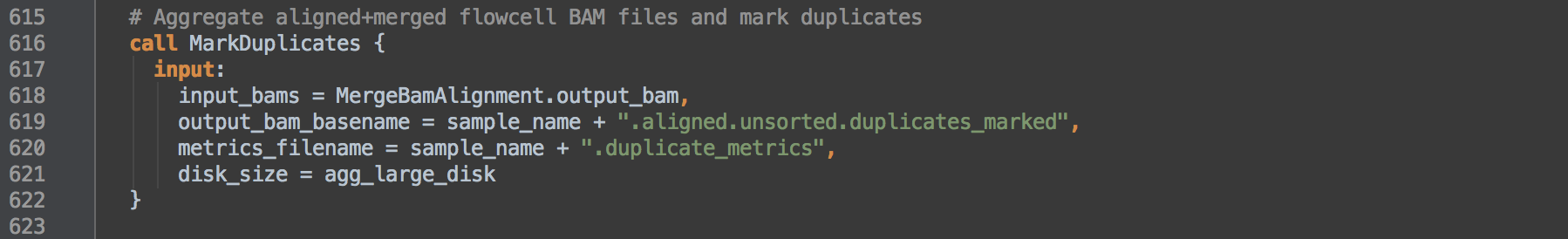
- 2.0: Calls task MarkDuplicates to accomplish two aims. First, since MarkDuplicates is given all the files output by the MergeBamAlignment task, and these by design all belong to the same sample, we effectively aggregate the starting BAMs into a single sample-level BAM. Second, MarkDuplicates flags reads it determines are from duplicate inserts with the 0x400 bitwise SAM flag. Because MarkDuplicates sees query-grouped read alignment records from the output of [1.3], it will also mark as duplicate the unmapped mates and supplementary alignments within the duplicate set.
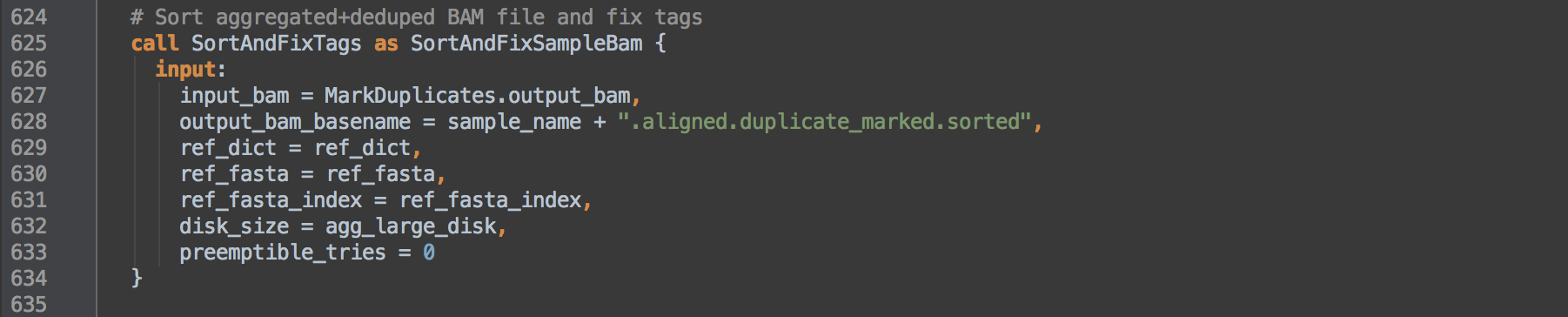
- 2.1: Calls task SortAndFixTags and renames it as SortAndFixSampleBam to differentiate outputs from those of [1.4] that calls the same task. This task coordinate sorts and indexes the alignments and fixes the
NM and UQ tags whose calculations depend on coordinate sort order. Resulting data go on to step [3].
[2.0]
[2.1]
3. Base quality score recalibration
This step creates intervals for scattering, performs BQSR, merges back the scattered results into a single file and finally compresses the BAM to CRAM format.
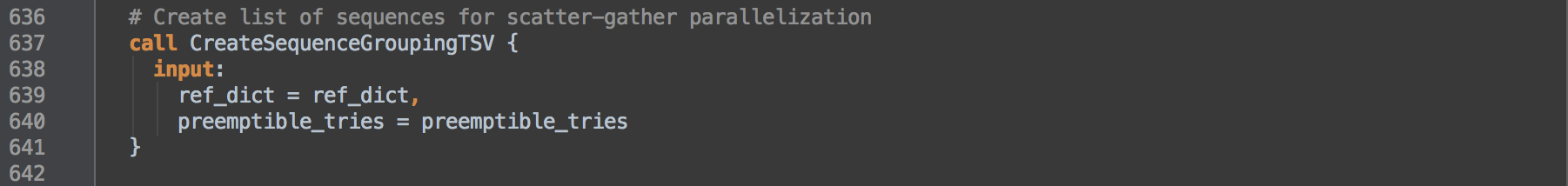
- 3.0: Calls task CreateSequenceGroupingTSV to create intervals from the reference genome's
.dict dictionary for subsequent use in boxes [3.1] and [3.2].
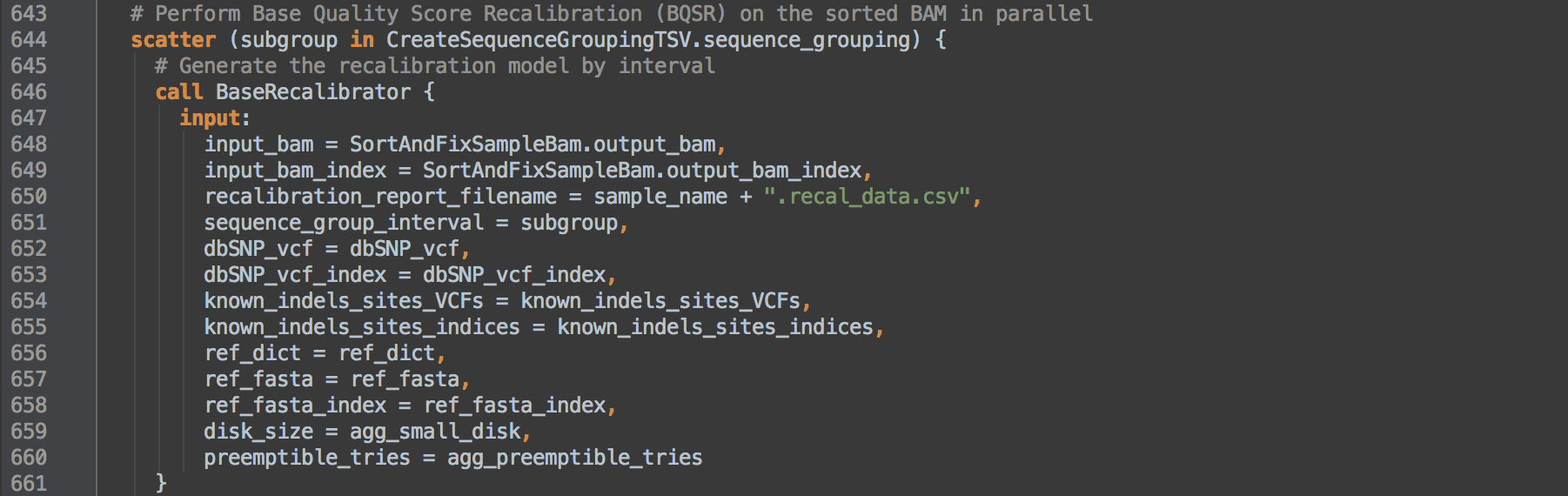
- 3.1: L644 defines scatter intervals as those created in box [3.0] to apply here and for [3.2]. Calls task BaseRecalibrator to act on each interval of the BAM from [2.1] and results in a recalibration table per interval.
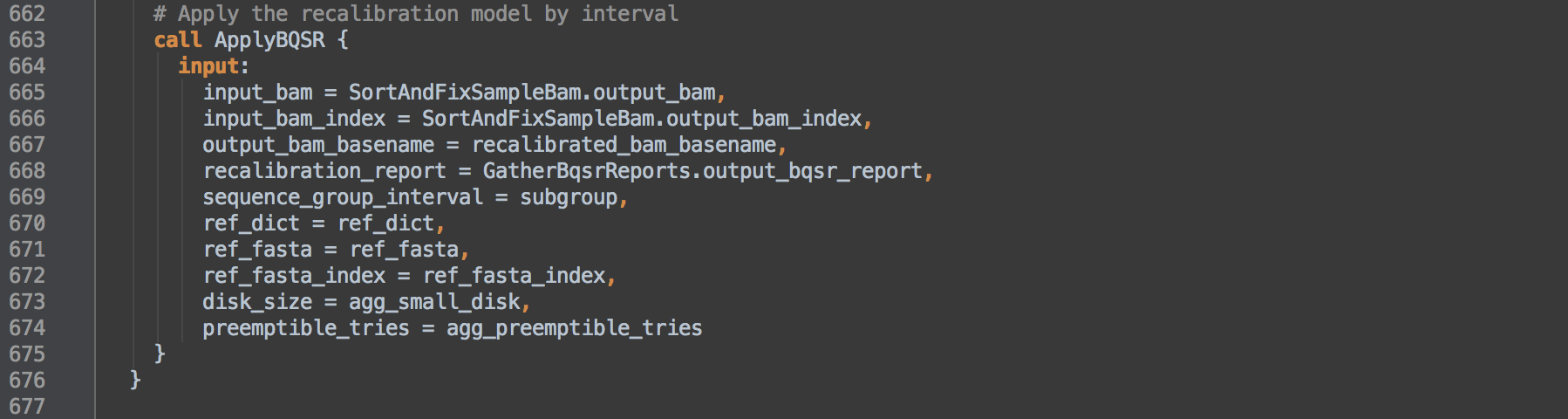
- 3.2: Calls task ApplyBQSR to use the
GatherBqsrReports.output_bqsr_report from [3.3] and apply the recalibration to the BAM from [2.1] per interval defined by [3.0]. Each resulting recalibrated BAM will contain alignment records from the specified interval including unmapped reads from singly mapping pairs. These unmapped records retain SAM alignment information, e.g. mapping contig and coordinate information, but have an asterisk * in the CIGAR field.
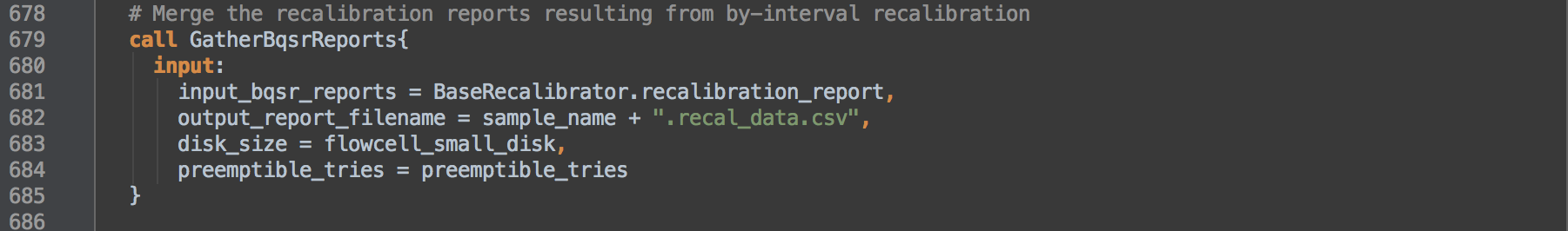
- 3.3: Calls task GatherBqsrReports to consolidate the per interval recalibration tables into a single recalibration table whose sums reflect the consolidated data.
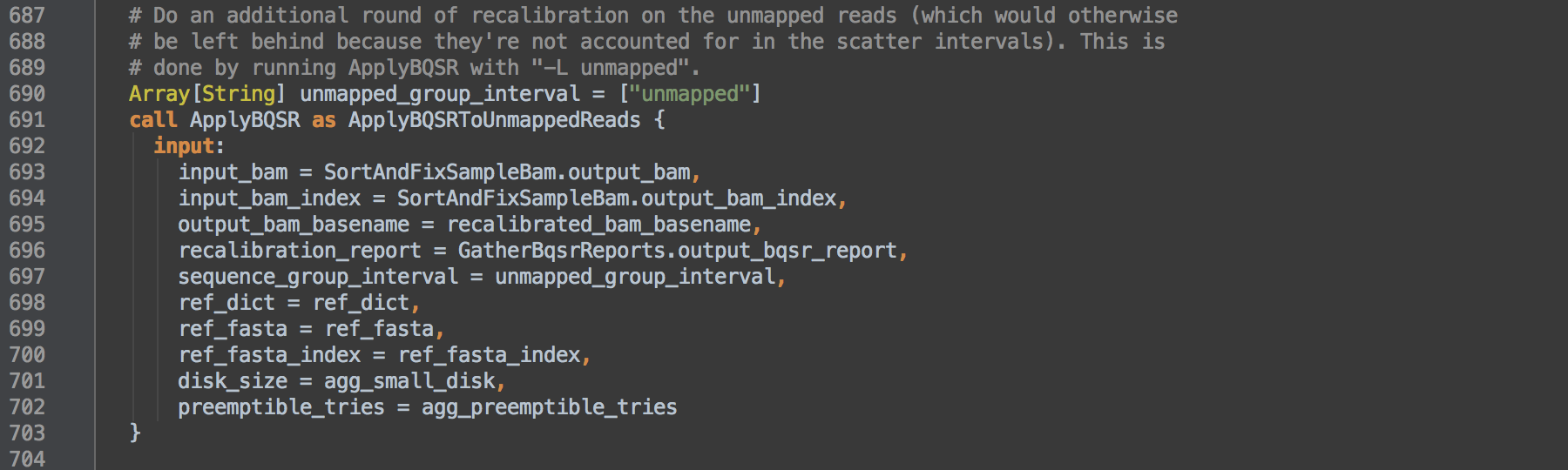
- 3.4: Calls task ApplyBQSR and uses task aliasing to rename it ApplyBQSRToUnmappedReads. The consequence of this is that the workflow can then differentiate outputs from those of [3.2]. The step takes as input the BAM from the SortAndFixSampleBam task [2.1] and L697 shows this command runs on unmapped SAM records. These are read pairs the aligner could not map and reads MergeBamAlignment unmapped as contaminants in [1.3] that are at the end of a coordinate-sorted BAM file. The resulting recalibrated BAM contains only such unmapped alignment records.
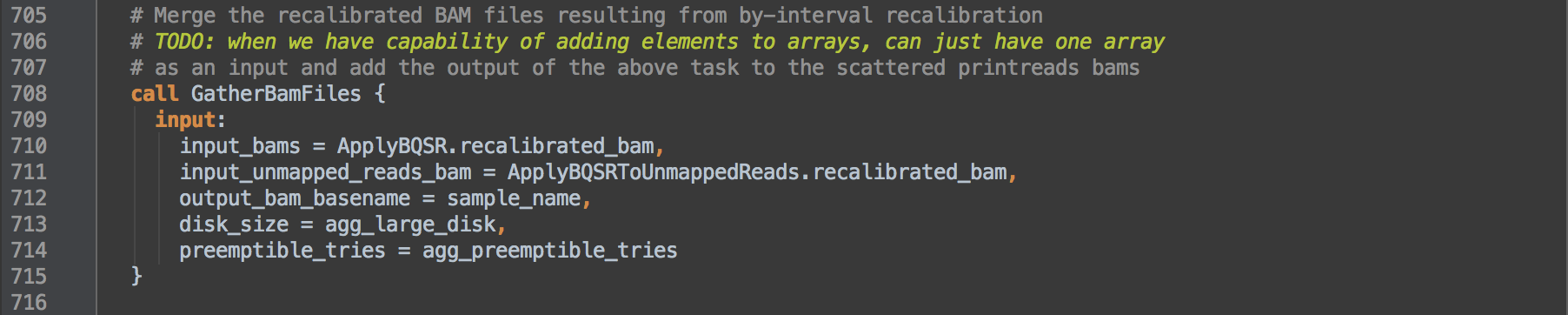
- 3.5: Calls task GatherBamFiles to concatenate the recalibrated BAMs from [3.2] and [3.4], in order, into a single indexed BAM that retains the header from the first BAM. Resulting data go onto step [4].
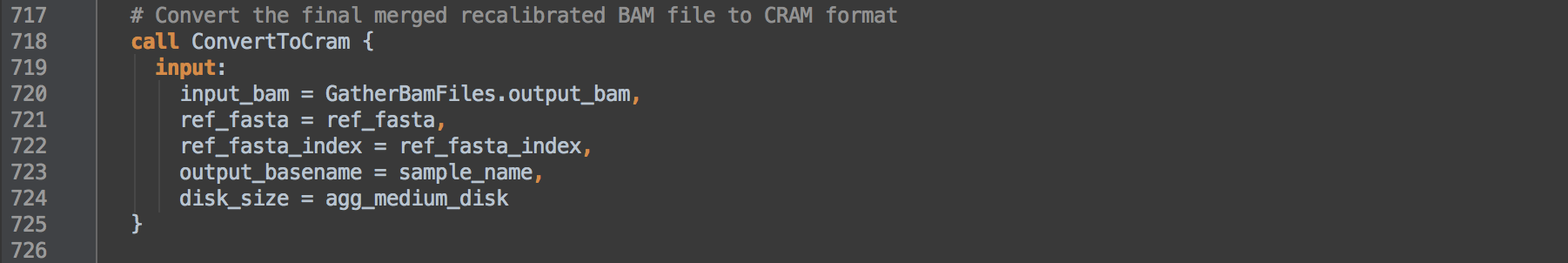
- 3.6: Calls task ConvertToCram to compress the BAM further in to reference-dependent indexed CRAM format.
[3.0]
[3.1]
[3.2]
[3.3]
[3.4]
[3.5]
[3.6]
back to top
4. Call SNP and INDEL variants with HaplotypeCaller
This final step uses HaplotypeCaller to call variants over intervals then merges data into a GVCF for the sample, the final output of the workflow.
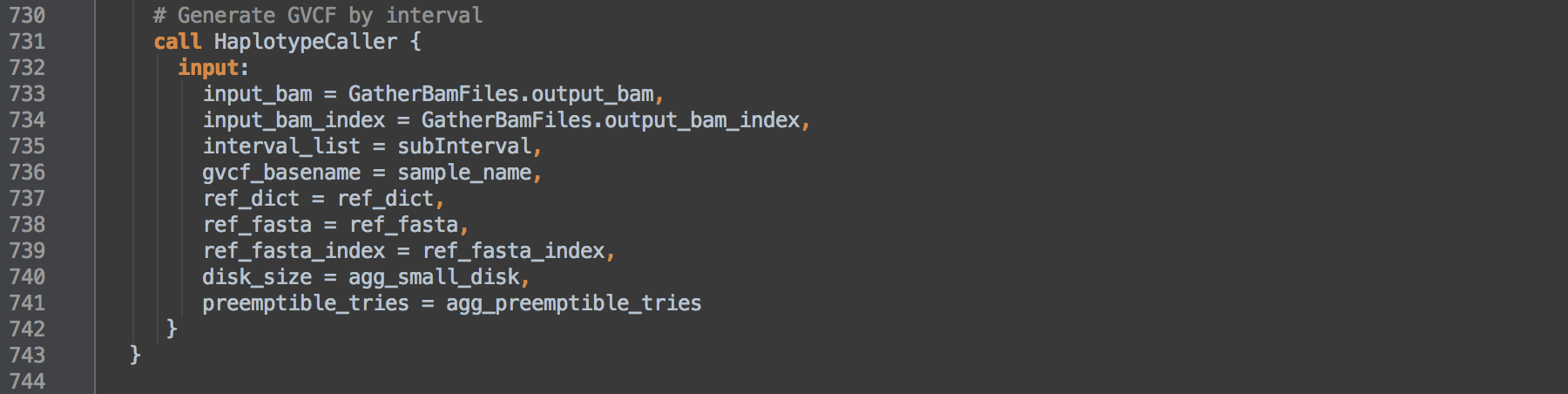
- 4.0: Uses scatter intervals defined within the JSON file under
scattered_calling_intervals (L728). We use only the primary assembly contigs of GRCh38, grouped into 50 intervals lists, to call variants. Within the GRCh38 intervals lists, the primary assembly's contigs are divided by contiguous regions between regions of Ns. The called task then uses this list of regions to parallelize the task via the -L ${interval_list} option.
▶︎ For this pipeline workflow's setup, fifty parallel processes makes sense for a genome of 3 billion basepairs. However, given the same setup, the 50-way split is overkill for a genome of 370 million basepairs as in the case of the pufferfish.
-
4.1: Calls task HaplotypeCaller to call SNP and INDEL variants on the BAM from [3.5] per interval and results in GVCF format variant calls files per interval.
-
4.2: Calls task GatherVCFs to merge the per interval GVCFs into one single-sample GVCF. The tool MergeVcfs concatenates the GVCFs in the order given by input_vcfs that by this WORKFLOW's design is ordered by contig.
- 4.3: Defines files that copy to an output directory if given an OPTIONS JSON file that defines the output directory. If you omit the OPTIONS JSON or omit defining the outputs directory in the OPTIONS JSON, then the workflow skips this step.
[4.0]

[4.1]
[4.2]
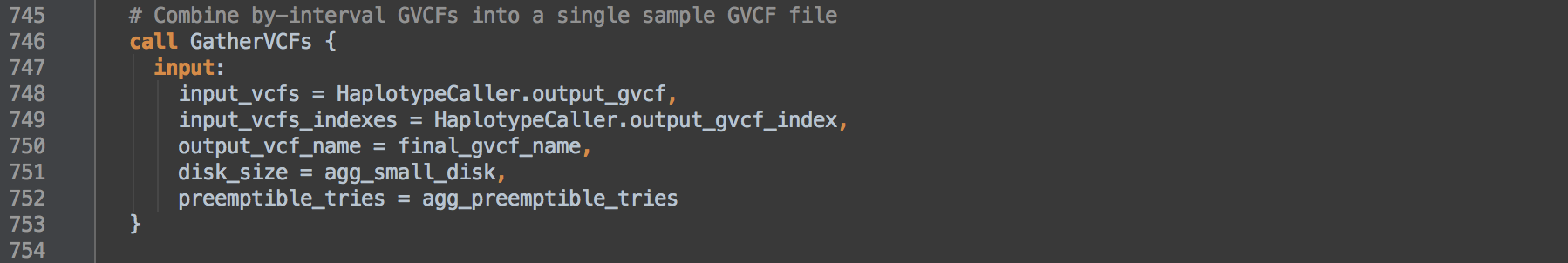
[4.3]
back to top
TASK DEFINITIONS
GetBwaVersion
This task obtains the version of BWA to later notate within the BAM program group (@PG) line.
# Get version of BWA
task GetBwaVersion {
command {
/usr/gitc/bwa 2>&1 | \
grep -e '^Version' | \
sed 's/Version: //'
}
runtime {
docker: "broadinstitute/genomes-in-the-cloud:2.2.3-1469027018"
memory: "1 GB"
}
output {
String version = read_string(stdout())
}
}
SamToFastqAndBwaMem
 The input to this task is an unaligned queryname-sorted BAM and the output is an aligned query-grouped BAM. This step pipes three processes: (i) conversion of BAM to FASTQ reads, (ii) [alternate-contig-aware alignment with BWA-MEM and (iii) conversion of SAM to BAM reads. BWA-MEM requires FASTQ reads as input and produces SAM format reads. This task maps the reads using the BWA command defined as a string variable and in this workflow this string is defined in [0.1].
The input to this task is an unaligned queryname-sorted BAM and the output is an aligned query-grouped BAM. This step pipes three processes: (i) conversion of BAM to FASTQ reads, (ii) [alternate-contig-aware alignment with BWA-MEM and (iii) conversion of SAM to BAM reads. BWA-MEM requires FASTQ reads as input and produces SAM format reads. This task maps the reads using the BWA command defined as a string variable and in this workflow this string is defined in [0.1].
The alt-aware alignment depends on use of GRCh38 as the reference, the versions 0.7.13+ of BWA and the presence of BWA's ALT index from bwa-kit. If the ref_alt ALT index has no content or is not present, then the script exits with an exit 1 error. What this means is that this task is only compatible with a reference with ALT contigs and it only runs in an alt-aware manner.
# Read unmapped BAM, convert on-the-fly to FASTQ and stream to BWA MEM for alignment
task SamToFastqAndBwaMem {
File input_bam
String bwa_commandline
String output_bam_basename
File ref_fasta
File ref_fasta_index
File ref_dict
# This is the .alt file from bwa-kit (https://github.com/lh3/bwa/tree/master/bwakit),
# listing the reference contigs that are "alternative".
File ref_alt
File ref_amb
File ref_ann
File ref_bwt
File ref_pac
File ref_sa
Int disk_size
Int preemptible_tries
command <<<
set -o pipefail
# set the bash variable needed for the command-line
bash_ref_fasta=${ref_fasta}
# if ref_alt has data in it,
if [ -s ${ref_alt} ]; then
java -Xmx3000m -jar /usr/gitc/picard.jar \
SamToFastq \
INPUT=${input_bam} \
FASTQ=/dev/stdout \
INTERLEAVE=true \
NON_PF=true | \
/usr/gitc/${bwa_commandline} /dev/stdin - 2> >(tee ${output_bam_basename}.bwa.stderr.log >&2) | \
samtools view -1 - > ${output_bam_basename}.bam && \
grep -m1 "read .* ALT contigs" ${output_bam_basename}.bwa.stderr.log | \
grep -v "read 0 ALT contigs"
# else ref_alt is empty or could not be found
else
exit 1;
fi
>>>
runtime {
docker: "broadinstitute/genomes-in-the-cloud:2.2.3-1469027018"
memory: "14 GB"
cpu: "16"
disks: "local-disk " + disk_size + " HDD"
preemptible: preemptible_tries
}
output {
File output_bam = "${output_bam_basename}.bam"
File bwa_stderr_log = "${output_bam_basename}.bwa.stderr.log"
}
}
MergeBamAlignment
This step takes an unmapped BAM and the aligned BAM and merges information from each. Reads, sequence and quality information and meta information from the unmapped BAM merge with the alignment information in the aligned BAM. The BWA version the script obtains from task GetBwaVersion is used here in the program group (@PG) bwamem. What is imperative for this step, that is implied by the script, is that the sort order of the unmapped and aligned BAMs are identical, i.e. query-group sorted. The BWA-MEM alignment step outputs reads in exactly the same order as they are input and so groups mates, secondary and supplementary alignments together for a given read name. The merging step requires both files maintain this ordering and will produce a final merged BAM in the same query-grouped order given the SORT_ORDER="unsorted" parameter. This has implications for how the MarkDuplicates task will flag duplicate sets.
Because the ATTRIBUTES_TO_RETAIN option is set to X0, any aligner-specific tags that are literally X0 will carryover to the merged BAM. BWA-MEM does not output such a tag but does output XS and XA tags for suboptimal alignment score and alternative hits, respectively. However, these do not carryover into the merged BAM. Merging retains certain tags from either input BAM (RG, SA, MD, NM, AS and OQ if present), replaces the PG tag as the command below defines and adds new tags (MC, MQ and FT).
▶︎ Note the NM tag values will be incorrect at this point and the UQ tag is absent. Update and addition of these are dependent on coordinate sort order. Specifically, the script uses a separate SortAndFixTags task to fix NM tags and add UQ tags.
The UNMAP_CONTAMINANT_READS=true option applies to likely cross-species contamination, e.g. bacterial contamination. MergeBamAlignment identifies reads that are (i) softclipped on both ends and (ii) map with less than 32 basepairs as contaminant. For a similar feature in GATK, see OverclippedReadFilter. If MergeBamAlignment determines a read is contaminant, then the mate is also considered contaminant. MergeBamAlignment unmaps the pair of reads by (i) setting the 0x4 flag bit, (ii) replacing column 3's contig name with an asterisk *, (iii) replacing columns 4 and 5 (POS and MAPQ) with zeros, and (iv) adding the FT tag to indicate the reason for unmapping the read, e.g. FT:Z:Cross-species contamination. The records retain their CIGAR strings. Note other processes also use the FT tag, e.g. to indicate reasons for setting the QCFAIL 0x200 flag bit, and will use different tag descriptions.
# Merge original input uBAM file with BWA-aligned BAM file
task MergeBamAlignment {
File unmapped_bam
String bwa_commandline
String bwa_version
File aligned_bam
String output_bam_basename
File ref_fasta
File ref_fasta_index
File ref_dict
Int disk_size
Int preemptible_tries
command {
# set the bash variable needed for the command-line
bash_ref_fasta=${ref_fasta}
java -Xmx3000m -jar /usr/gitc/picard.jar \
MergeBamAlignment \
VALIDATION_STRINGENCY=SILENT \
EXPECTED_ORIENTATIONS=FR \
ATTRIBUTES_TO_RETAIN=X0 \
ALIGNED_BAM=${aligned_bam} \
UNMAPPED_BAM=${unmapped_bam} \
OUTPUT=${output_bam_basename}.bam \
REFERENCE_SEQUENCE=${ref_fasta} \
PAIRED_RUN=true \
SORT_ORDER="unsorted" \
IS_BISULFITE_SEQUENCE=false \
ALIGNED_READS_ONLY=false \
CLIP_ADAPTERS=false \
MAX_RECORDS_IN_RAM=2000000 \
ADD_MATE_CIGAR=true \
MAX_INSERTIONS_OR_DELETIONS=-1 \
PRIMARY_ALIGNMENT_STRATEGY=MostDistant \
PROGRAM_RECORD_ID="bwamem" \
PROGRAM_GROUP_VERSION="${bwa_version}" \
PROGRAM_GROUP_COMMAND_LINE="${bwa_commandline}" \
PROGRAM_GROUP_NAME="bwamem" \
UNMAP_CONTAMINANT_READS=true
}
runtime {
docker: "broadinstitute/genomes-in-the-cloud:2.2.3-1469027018"
memory: "3500 MB"
cpu: "1"
disks: "local-disk " + disk_size + " HDD"
preemptible: preemptible_tries
}
output {
File output_bam = "${output_bam_basename}.bam"
}
}
MarkDuplicates
This task flags duplicate reads. Because the input is query-group-sorted, MarkDuplicates flags with the 0x400 bitwise SAM flag duplicate primary alignments as well as the duplicate set's secondary and supplementary alignments. Also, for singly mapping mates, duplicate flagging extends to cover unmapped mates. These extensions are features that are only available to query-group-sorted BAMs.
This command uses the ASSUME_SORT_ORDER="queryname" parameter to tell the tool the sort order to expect. Within the context of this workflow, at the point this task is called, we will have avoided any active sorting that would label the BAM header. We know that our original flowcell BAM is queryname-sorted and that BWA-MEM maintains this order to give us query-grouped alignments.
The OPTICAL_DUPLICATE_PIXEL_DISTANCE of 2500 is set for Illumina sequencers that use patterned flowcells to estimate the number of sequencer duplicates. Sequencer duplicates are a subspecies of the duplicates that the tool flags. The Illumina HiSeq X and HiSeq 4000 platforms use patterened flowcells. If estimating library complexity (see section Duplicate metrics in brief) is important to you, then adjust the OPTICAL_DUPLICATE_PIXEL_DISTANCE appropriately for your sequencer platform.
Finally, in this task and others, we produce an MD5 file with the CREATE_MD5_FILE=true option. This creates a 128-bit hash value using the MD5 algorithm that is to files much like a fingerprint is to an individual. Compare MD5 values to verify data integrity, e.g. after moving or copying large files.
# Mark duplicate reads to avoid counting non-independent observations
task MarkDuplicates {
Array[File] input_bams
String output_bam_basename
String metrics_filename
Int disk_size
# Task is assuming query-sorted input so that the Secondary and Supplementary reads get marked correctly
# This works because the output of BWA is query-grouped, and thus so is the output of MergeBamAlignment.
# While query-grouped isn't actually query-sorted, it's good enough for MarkDuplicates with ASSUME_SORT_ORDER="queryname"
command {
java -Xmx4000m -jar /usr/gitc/picard.jar \
MarkDuplicates \
INPUT=${sep=' INPUT=' input_bams} \
OUTPUT=${output_bam_basename}.bam \
METRICS_FILE=${metrics_filename} \
VALIDATION_STRINGENCY=SILENT \
OPTICAL_DUPLICATE_PIXEL_DISTANCE=2500 \
ASSUME_SORT_ORDER="queryname"
CREATE_MD5_FILE=true
}
runtime {
docker: "broadinstitute/genomes-in-the-cloud:2.2.3-1469027018"
memory: "7 GB"
disks: "local-disk " + disk_size + " HDD"
}
output {
File output_bam = "${output_bam_basename}.bam"
File duplicate_metrics = "${metrics_filename}"
}
}
SortAndFixTags
This task (i) sorts reads by coordinate and then (ii) corrects the NM tag values, adds UQ tags and indexes a BAM. The task pipes the two commands. First, SortSam sorts the records by genomic coordinate using the SORT_ORDER="coordinate" option. Second, SetNmAndUqTags calculates and fixes the UQ and NM tag values in the BAM. Because CREATE_INDEX=true, SetNmAndUqTags creates the .bai index. Again, we create an MD5 file with the CREATE_MD5_FILE=true option.
As mentioned in the MergeBamAlignment task, tag values dependent on coordinate-sorted records require correction in this separate task given this workflow maintains query-group ordering through the pre-processing steps.
# Sort BAM file by coordinate order and fix tag values for NM and UQ
task SortAndFixTags {
File input_bam
String output_bam_basename
File ref_dict
File ref_fasta
File ref_fasta_index
Int disk_size
Int preemptible_tries
command {
java -Xmx4000m -jar /usr/gitc/picard.jar \
SortSam \
INPUT=${input_bam} \
OUTPUT=/dev/stdout \
SORT_ORDER="coordinate" \
CREATE_INDEX=false \
CREATE_MD5_FILE=false | \
java -Xmx500m -jar /usr/gitc/picard.jar \
SetNmAndUqTags \
INPUT=/dev/stdin \
OUTPUT=${output_bam_basename}.bam \
CREATE_INDEX=true \
CREATE_MD5_FILE=true \
REFERENCE_SEQUENCE=${ref_fasta}
}
runtime {
docker: "broadinstitute/genomes-in-the-cloud:2.2.3-1469027018"
disks: "local-disk " + disk_size + " HDD"
cpu: "1"
memory: "5000 MB"
preemptible: preemptible_tries
}
output {
File output_bam = "${output_bam_basename}.bam"
File output_bam_index = "${output_bam_basename}.bai"
File output_bam_md5 = "${output_bam_basename}.bam.md5"
}
}
CreateSequenceGroupingTSV
This task uses a python script written as a single command using heredoc syntax to create a list of contig groupings. The workflow uses the intervals to scatter the base quality recalibration step [3] that calls on BaseRecalibrator and ApplyBQSR tasks.
This workflow specifically uses Python v2.7.
The input to the task is the reference .dict dictionary that lists contigs. The code takes the information provided by the SN and LN tags of each @SQ line in the dictionary to pair the information in a tuple list. The SN tag names a contig while the LN tag measures the contig length. This list is ordered by descending contig length.
The contig groupings this command creates is in WDL array format where each line represents a group and each group's members are tab-separated. The command adds contigs to each group from the previously length-sorted list in descending order and caps the sum of member lengths by the first contig's sequence length (the longest contig). This has the effect of somewhat evenly distributing sequence per group. For GRCh38, CreateSequenceGroupingTSV-stdout.log shows 18 such groups.
As the code adds contig names to groups, it adds a :1+ to the end of each name. This is to protect the names from downstream tool behavior that removes elements after the last : within a contig name. GRCh38 introduces contig names that include :s and removing the last element make certain contigs indistinguishable from others. With this appendage, we preserve the original contig names through downstream processes. GATK v3.5 and prior versions require this addition.
# Generate sets of intervals for scatter-gathering over chromosomes
task CreateSequenceGroupingTSV {
File ref_dict
Int preemptible_tries
# Use python to create the Sequencing Groupings used for BQSR and PrintReads Scatter. It outputs to stdout
# where it is parsed into a wdl Array[Array[String]]
# e.g. [["1"], ["2"], ["3", "4"], ["5"], ["6", "7", "8"]]
command <<<
python <<CODE
with open("${ref_dict}", "r") as ref_dict_file:
sequence_tuple_list = []
longest_sequence = 0
for line in ref_dict_file:
if line.startswith("@SQ"):
line_split = line.split("\t")
# (Sequence_Name, Sequence_Length)
sequence_tuple_list.append((line_split[1].split("SN:")[1], int(line_split[2].split("LN:")[1])))
longest_sequence = sorted(sequence_tuple_list, key=lambda x: x[1], reverse=True)[0][1]
# We are adding this to the intervals because hg38 has contigs named with embedded colons and a bug in GATK strips off
# the last element after a :, so we add this as a sacrificial element.
hg38_protection_tag = ":1+"
# initialize the tsv string with the first sequence
tsv_string = sequence_tuple_list[0][0] + hg38_protection_tag
temp_size = sequence_tuple_list[0][1]
for sequence_tuple in sequence_tuple_list[1:]:
if temp_size + sequence_tuple[1] <= longest_sequence:
temp_size += sequence_tuple[1]
tsv_string += "\t" + sequence_tuple[0] + hg38_protection_tag
else:
tsv_string += "\n" + sequence_tuple[0] + hg38_protection_tag
temp_size = sequence_tuple[1]
print tsv_string
CODE
>>>
runtime {
docker: "python:2.7"
memory: "2 GB"
preemptible: preemptible_tries
}
output {
Array[Array[String]] sequence_grouping = read_tsv(stdout())
}
}
BaseRecalibrator
The task runs BaseRecalibrator to detect errors made by the sequencer in estimating base quality scores. BaseRecalibrator builds a model of covariation from mismatches in the alignment data while excluding known variant sites and creates a recalibration report for use in the next step. The engine parameter --useOriginalQualities asks BaseRecalibrator to use original sequencer-produced base qualities stored in the OQ tag if present or otherwise use the standard QUAL score. The known sites files should include sites of known common SNPs and INDELs.
This task runs per interval grouping defined by each -L option. The sep in -L ${sep=" -L " sequence_group_interval} ensures each interval in the _sequence_groupinterval list is given by the command.
# Generate Base Quality Score Recalibration (BQSR) model
task BaseRecalibrator {
File input_bam
File input_bam_index
String recalibration_report_filename
Array[String] sequence_group_interval
File dbSNP_vcf
File dbSNP_vcf_index
Array[File] known_indels_sites_VCFs
Array[File] known_indels_sites_indices
File ref_dict
File ref_fasta
File ref_fasta_index
Int disk_size
Int preemptible_tries
command {
java -XX:GCTimeLimit=50 -XX:GCHeapFreeLimit=10 -XX:+PrintFlagsFinal \
-XX:+PrintGCTimeStamps -XX:+PrintGCDateStamps -XX:+PrintGCDetails \
-Xloggc:gc_log.log -Dsamjdk.use_async_io=false -Xmx4000m \
-jar /usr/gitc/GATK4.jar \
BaseRecalibrator \
-R ${ref_fasta} \
-I ${input_bam} \
--useOriginalQualities \
-O ${recalibration_report_filename} \
-knownSites ${dbSNP_vcf} \
-knownSites ${sep=" -knownSites " known_indels_sites_VCFs} \
-L ${sep=" -L " sequence_group_interval}
}
runtime {
docker: "broadinstitute/genomes-in-the-cloud:2.2.3-1469027018"
memory: "6 GB"
disks: "local-disk " + disk_size + " HDD"
preemptible: preemptible_tries
}
output {
File recalibration_report = "${recalibration_report_filename}"
#this output is only for GOTC STAGING to give some GC statistics to the GATK4 team
#File gc_logs = "gc_log.log"
}
}
GatherBqsrReports
This task consolidates the recalibration reports from each sequence group interval into a single report using GatherBqsrReports.
# Combine multiple recalibration tables from scattered BaseRecalibrator runs
task GatherBqsrReports {
Array[File] input_bqsr_reports
String output_report_filename
Int disk_size
Int preemptible_tries
command {
java -Xmx3000m -jar /usr/gitc/GATK4.jar \
GatherBQSRReports \
-I ${sep=' -I ' input_bqsr_reports} \
-O ${output_report_filename}
}
runtime {
docker: "broadinstitute/genomes-in-the-cloud:2.2.3-1469027018"
memory: "3500 MB"
disks: "local-disk " + disk_size + " HDD"
preemptible: preemptible_tries
}
output {
File output_bqsr_report = "${output_report_filename}"
}
}
ApplyBQSR
The task uses ApplyBQSR and the recalibration report to correct base quality scores in the BAM. Again, using parallelization, this task applies recalibration for the sequence intervals defined with -L. A resulting recalibrated BAM will contain only reads for the intervals in the applied intervals list.
# Apply Base Quality Score Recalibration (BQSR) model
task ApplyBQSR {
File input_bam
File input_bam_index
String output_bam_basename
File recalibration_report
Array[String] sequence_group_interval
File ref_dict
File ref_fasta
File ref_fasta_index
Int disk_size
Int preemptible_tries
command {
java -XX:+PrintFlagsFinal -XX:+PrintGCTimeStamps -XX:+PrintGCDateStamps \
-XX:+PrintGCDetails -Xloggc:gc_log.log -Dsamjdk.use_async_io=false \
-XX:GCTimeLimit=50 -XX:GCHeapFreeLimit=10 -Xmx3000m \
-jar /usr/gitc/GATK4.jar \
ApplyBQSR \
--createOutputBamMD5 \
--addOutputSAMProgramRecord \
-R ${ref_fasta} \
-I ${input_bam} \
--useOriginalQualities \
-O ${output_bam_basename}.bam \
-bqsr ${recalibration_report} \
-SQQ 10 -SQQ 20 -SQQ 30 -SQQ 40 \
--emit_original_quals \
-L ${sep=" -L " sequence_group_interval}
}
runtime {
docker: "broadinstitute/genomes-in-the-cloud:2.2.3-1469027018"
memory: "3500 MB"
disks: "local-disk " + disk_size + " HDD"
preemptible: preemptible_tries
}
output {
File recalibrated_bam = "${output_bam_basename}.bam"
File recalibrated_bam_checksum = "${output_bam_basename}.bam.md5"
#this output is only for GOTC STAGING to give some GC statistics to the GATK4 team
#File gc_logs = "gc_log.log"
}
}
GatherBamFiles
This task concatenates provided BAMs in order, into a single BAM and retains the header of the first file. For this pipeline, this includes the recalibrated sequence grouped BAMs and the recalibrated unmapped reads BAM. For GRCh38, this makes 19 BAM files that the task concatenates together. The resulting BAM is already in coordinate-sorted order. The task creates a new sequence index and MD5 file for the concatenated BAM.
# Combine multiple recalibrated BAM files from scattered ApplyRecalibration runs
task GatherBamFiles {
Array[File] input_bams
File input_unmapped_reads_bam
String output_bam_basename
Int disk_size
Int preemptible_tries
command {
java -Xmx2000m -jar /usr/gitc/picard.jar \
GatherBamFiles \
INPUT=${sep=' INPUT=' input_bams} \
INPUT=${input_unmapped_reads_bam} \
OUTPUT=${output_bam_basename}.bam \
CREATE_INDEX=true \
CREATE_MD5_FILE=true
}
runtime {
docker: "broadinstitute/genomes-in-the-cloud:2.2.3-1469027018"
memory: "3 GB"
disks: "local-disk " + disk_size + " HDD"
preemptible: preemptible_tries
}
output {
File output_bam = "${output_bam_basename}.bam"
File output_bam_index = "${output_bam_basename}.bai"
File output_bam_md5 = "${output_bam_basename}.bam.md5"
}
}
ConvertToCram
This task compresses a BAM to an even smaller CRAM format using the -C option of Samtools. The task then indexes the CRAM and renames it from {basename}.cram.crai to {basename}.crai. CRAM is a new format and tools are actively refining features for compatibility. Make sure your tool chain is compatible with CRAM before deleting BAMs. Be aware when using CRAMs that you will have to specify the identical reference genome, not just equivalent reference, with matching MD5 hashes for each contig. These can differ if the capitalization of reference sequences differ.
# Convert BAM file to CRAM format
task ConvertToCram {
File input_bam
File ref_fasta
File ref_fasta_index
String output_basename
Int disk_size
# Note that we are not activating pre-emptible instances for this step yet,
# but we should if it ends up being fairly quick
command <<<
samtools view -C -T ${ref_fasta} ${input_bam} | \
tee ${output_basename}.cram | \
md5sum > ${output_basename}.cram.md5 && \
samtools index ${output_basename}.cram && \
mv ${output_basename}.cram.crai ${output_basename}.crai
>>>
runtime {
docker: "broadinstitute/genomes-in-the-cloud:2.2.3-1469027018"
memory: "3 GB"
cpu: "1"
disks: "local-disk " + disk_size + " HDD"
}
output {
File output_cram = "${output_basename}.cram"
File output_cram_index = "${output_basename}.crai"
File output_cram_md5 = "››${output_basename}.cram.md5"
}
}
HaplotypeCaller
This task runs HaplotypeCaller on the recalibrated BAM for given intervals and produces variant calls in GVCF format. HaplotypeCaller reassembles and realign reads around variants and calls genotypes and genotype likelihoods for single nucleotide polymorphism (SNP) and insertion and deletion (INDELs) variants. Proximal variants are phased. The resulting file is a GZ compressed file, a valid VCF format file with extension .vcf.gz, containing variants for the given interval.
- The WORKFLOW's step 4 defines any parallelization.
The -ERC GVCF or emit reference confidence mode activates two GVCF features. First, for each variant call, we now include a symbolic <NON_REF> non-reference allele. Second, for non-variant regions, we now include <NON_REF> summary blocks as calls.
- The
--max_alternate_alleles is set to three for performance optimization. This does not limit the alleles that are genotyped, only the number of alleles that HaplotypeCaller emits.
- Because this WORKFLOW's naming convention does not use the
.g.vcf extension, we must specify -variant_index_parameter 128000 and -variant_index_type LINEAR to set the correct index strategy for the output GVCF. See Article#3893 for details.
- The command invokes an additional read, the OverclippedReadFilter, with
--read_filter OverclippedRead that removes reads that are likely from foreign contaminants, e.g. bacterial contamination. The filter define such reads as those that align with less than 30 basepairs and are softclipped on both ends of the read. This option is similar to the MergeBamAlignment task's UNMAP_CONTAMINANT_READS=true option that unmaps contaminant reads less than 32 basepairs.
# Call variants on a single sample with HaplotypeCaller to produce a GVCF
task HaplotypeCaller {
File input_bam
File input_bam_index
File interval_list
String gvcf_basename
File ref_dict
File ref_fasta
File ref_fasta_index
Float? contamination
Int disk_size
Int preemptible_tries
# tried to find lowest memory variable where it would still work, might change once tested on JES
command {
java -XX:GCTimeLimit=50 -XX:GCHeapFreeLimit=10 -Xmx8000m \
-jar /usr/gitc/GATK35.jar \
-T HaplotypeCaller \
-R ${ref_fasta} \
-o ${gvcf_basename}.vcf.gz \
-I ${input_bam} \
-L ${interval_list} \
-ERC GVCF \
--max_alternate_alleles 3 \
-variant_index_parameter 128000 \
-variant_index_type LINEAR \
-contamination ${default=0 contamination} \
--read_filter OverclippedRead
}
runtime {
docker: "broadinstitute/genomes-in-the-cloud:2.2.3-1469027018"
memory: "10 GB"
cpu: "1"
disks: "local-disk " + disk_size + " HDD"
preemptible: preemptible_tries
}
output {
File output_gvcf = "${gvcf_basename}.vcf.gz"
File output_gvcf_index = "${gvcf_basename}.vcf.gz.tbi"
}
}
GatherVCFs
The task uses MergeVcfs to combine multiple VCF files into a single VCF file and index.
# Combine multiple VCFs or GVCFs from scattered HaplotypeCaller runs
task GatherVCFs {
Array[File] input_vcfs
Array[File] input_vcfs_indexes
String output_vcf_name
Int disk_size
Int preemptible_tries
# using MergeVcfs instead of GatherVcfs so we can create indices
# WARNING 2015-10-28 15:01:48 GatherVcfs Index creation not currently supported when gathering block compressed VCFs.
command {
java -Xmx2g -jar /usr/gitc/picard.jar \
MergeVcfs \
INPUT=${sep=' INPUT=' input_vcfs} \
OUTPUT=${output_vcf_name}
}
output {
File output_vcf = "${output_vcf_name}"
File output_vcf_index = "${output_vcf_name}.tbi"
}
runtime {
docker: "broadinstitute/genomes-in-the-cloud:2.2.3-1469027018"
memory: "3 GB"
disks: "local-disk " + disk_size + " HDD"
preemptible: preemptible_tries
}
}
back to top